 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipLet’s take a look at one of the more irritating issues in LC/MS method development: signal loss. I’m not talking about signal attenuation, which can be much more convoluted to diagnose; but rather a complete loss of signal – nothing, not even a blip on your TIC. So where do we begin?
Last time we wrapped up tuning our analytes to the mass spectrometers optics.
Our method was up and running samples day-in and day-out. We took it offline for couple of weeks and when we return to run the same extraction, the same LC/MS method – even the same LC column – we get nothing. My +40-analyte panel was humming along, and after a brief three-week hiatus, I came back to run the same assay when the lights went out (figure 1). I had installed the column I was using for the assay, prepped fresh mobile phase and injection port wash solvent, all according to the SOP. Next, I prepped my standard cocktail (fresh) for a retention time check and made a direct injection. Looking at the TIC, it appeared as if I hadn’t even made an injection! I double-checked to make sure I didn’t accidentally inject my blank. Nope, I had the correct vial.
So what happened?
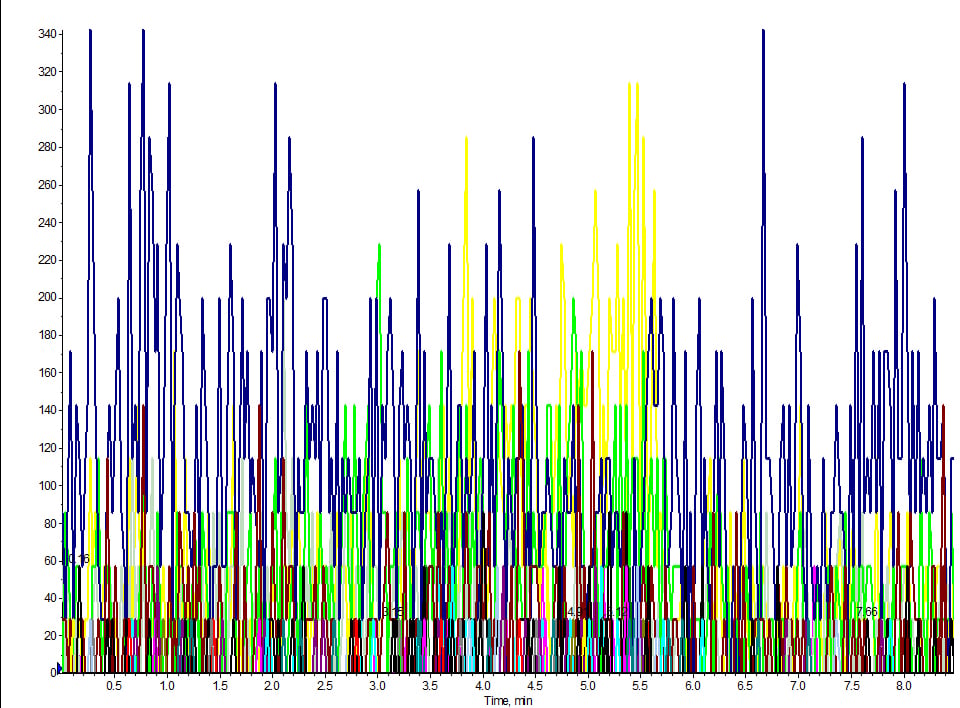 Figure 1. LC/MS MRM trace of direct injected standard cocktail at 100 ng/mL
Figure 1. LC/MS MRM trace of direct injected standard cocktail at 100 ng/mL
Fortunately, the complete loss signal typically indicates there’s a singular event that has managed to take our system off-line.
Where to start?
For the lucky few who have a service contract, you pick up the phone.
For the rest of us, we begin with the low-hanging fruit:
Is the issue with the extraction, the LC, or the MS?
The easiest task here is to remove the extraction from the equation and prep fresh standards. Next, I wanted to be sure the MS is functioning properly. I compare this approach to a car engine, that is, do I have all of the following, so my engine (my MS) is turning over:
- -spark (voltages for spray and optics)
- -air (nitrogen for nebulization/drying)
- -fuel (mobile phase)
The combination of all three (including heat) must be present to produce a stable ESI spray. We can look at the feedback values via the MS software, but not all manufactures allow this (a sardonic thanks to Sciex!). So, grab a flashlight or a laser pointer and focus either on the tip of the ESI needle, if you see a spray, chances are good the MS source is functioning. Indeed, in my case, I had a stable spray.
Because I had so many analytes, I decided to focus on one I knew functioned routinely: meprobamate. From here, I took the LC out of the equation, which left me with the MS optics to evaluate. I had a spray, so I took my standard cocktail and integrated my infusion pump directly to the source of the MS. If my spray was stable, I should be able to make droplets and, thus, see meprobamate in Q1 (figure 2).
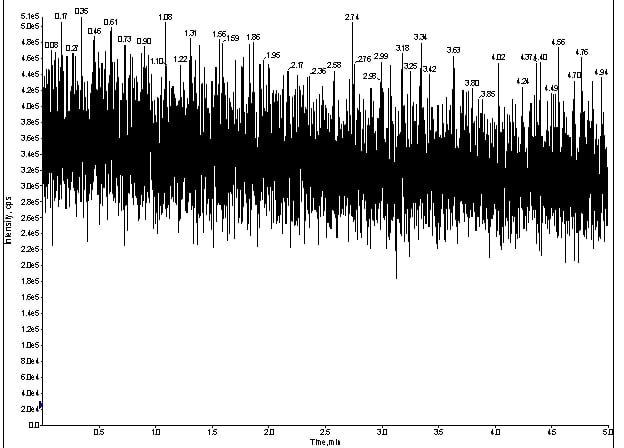
Figure 2. Q1 trace of for direct infusion of meprobamate without LC at 100ng/mL.
After infusing 100ng/mL of my cocktail, I could see the TIC produced by the Q1 spectra for meprobamate (figure 2).
At this stage, I knew my analytes were getting into the source, so my source parameters were fine.
The Q1 trace was smooth and uneventful, so I reconnected the LC flow, sans column, at [50:50] MBA/MPB and infused the same cocktail (figure 3).
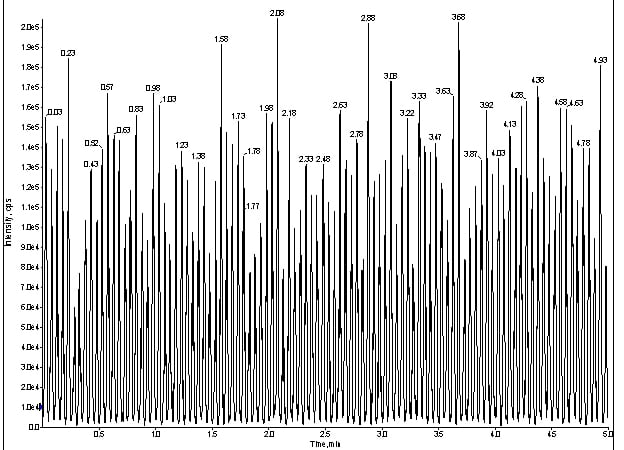
Figure 3. Q1 trace of for direct infusion of meprobamate with LC at 100ng/mL.
Interestingly, a reciprocating signal popped up in my Q1 trace (figure 3). At this point, I was not completely convinced this artifact was related to my missing peaks. Usually, the absence of peaks means loss of chromatography, so I re-installed the LC flow (with column) and monitored my infusion as before, but with the MRM for meprobamate (figure 4).
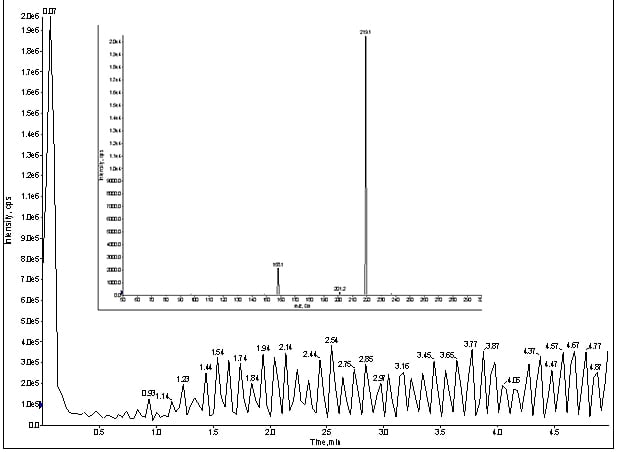
Figure 4. MRM trace of for direct infusion of meprobamate with LC & column, at 100ng/mL (Inset spectrum: MRM for meprobamate).
Once again, the reciprocating signal appeared once the LC connected to the system, but still no peaks.
At this point, I suspected the LC and I swapped columns, which proved to be fruitless as the same trace showed up as seen figure 4, so it was not the chromatography.
Nevertheless, the predictable nature of the MS signal when the LC was inline finally drew my attention to the one device whose sole objective is reciprocation: the pumps. Surely, I had purged them (I had) so perhaps another purge would help.
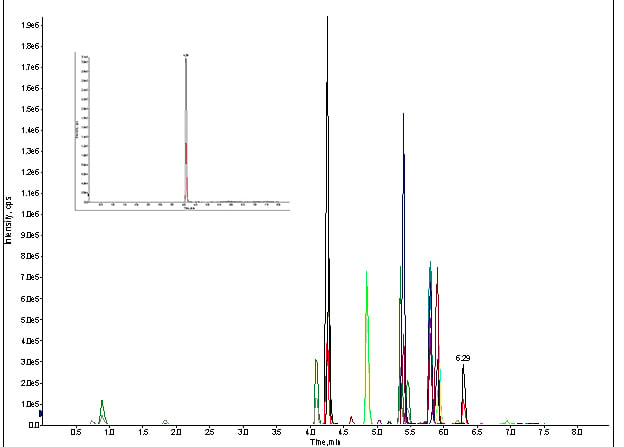
Figure 5. MRM trace of for direct injection of standard cocktail with LC & column, at 100ng/mL (Inset spectrum: extracted MRM for meprobamate).
It did. There were my peaks, but not for long. After a second injection, they were gone again.
So, what was happening?
It turns out I was losing the prime on my B pump (my organic phase) due to a large air pocket. The air pocket would not dislodge when the system attempted an auto-purge. Hence, I wasn’t getting any chromatography because there wasn’t any on-column solvation of my analytes. I primed the system again, and there were my peaks. Once that was manually removed, via syringe, my assay was up and running!
Next time my peaks pull a Houdini, I’ll be sure to manually purge my pumps.
What about the reverse scenario? Got peaks when you shouldn’t? Read more on our blog about troubleshooting this all-too-common problem.